ANALYSIS OF LNCRNAS AND CIRCRNAS IN GLYCINE MAX UNDER DROUGHT AND SALINE-ALKALINE STRESSES
Y. Dong, Y. Wang, G. Wang, N. Ahamd, L. Wang, Y. Wang, X. Zhang, X. Li, and H. Li*
College of Life Sciences, Engineering Research Center of Bioreactor and Pharmaceutical Development Ministry of Education, Jilin Agricultural University, Changchun 130118, China
*Corresponding Authour’s email: hyli99@163.com
ABSTRACT
Long noncoding-RNAs (LncRNAs) and circular RNAs (circRNAs) are the major types of noncoding RNAs (ncRNAs) known to be involved during diverse biological processes in eukaryotes. However, less attention has been given to the identification, overall characteristics and expression analysis of LncRNAs and circRNAs under abiotic stresses in plants.. In our study, strand-specific RNA-seq data revealed the identification of a total 2,020 lncRNAs, including 1,979 known and 41 novel ones, and 830 circRNAs in soybean. Overall, 118 and 47 lncRNAs showed significant changes in expression levels under drought stress and saline-alkaline stress, respectively. In addition, soybean lncRNAs indicated diverse expression patterns suggesting a positive correlation with many biological processes including photosynthesis during stress responses by regulating gene expression in a cis-/trans-actingmanner. Our findings further indicated that transcriptional biosynthesis pathways and RNA transcriptional regulation were positively regulated during stress conditions. Furthermore, the back-splicing sites of circRNAs were also confirmed. Collectively, our study identified a group of stress-responsive lncRNAs and circRNAs in soybean, highlighting their potential contributions to abiotic stress responses, which pave the way for future functional studies.
Keywords:Strand-specific RNA sequencing; Stress response; lncRNA; circRNA; Expression pattern;
http://doi.org/10.36899/JAPS.2022.3.0483
Published first online October 19. 2021
INTRODUCTION
Among the abiotic stresses that negatively affect the growth and development of crop plants, environmental factors are the most common inhibitors of yield(Bouain et al. 2019). Glycine max (soybean) is an economically important crop that is sensitive to abiotic stresses, particularly drought and saline-alkaline stresses(Kunert et al. 2016). This sensitivity means that there has been only limited success in breeding stress-resistant soybean cultivars. To overcome these limitations and increase the productivity of soybean, the molecular mechanisms of stress tolerance and their interaction with environmental factors should be better understood(Deinlein et al. 2014).
Noncoding RNAs are now recognized as crucial components of the stress response signaling networks that regulate plant defense systems. Long noncoding RNAs (lncRNAs) are defined as aheterogeneous group of noncoding RNAs whose transcripts are longer than 200 nucleotides (nt) (Kapranov et al. 2007). They do not contain any evolutionarily conserved open reading frames (Rivas et al. 2017). Previous studies have shown that they are expressed in a tissue- or stage-specific manner and their expression levels change in response to various environmental factors (Di et al. 2014; Liu et al. 2015; Qi et al. 2013; Rymarquis et al. 2008). The lncRNAs are functionally significant because they regulate RNA processing machinery in a cis or trans manner (Kung et al. 2013). Emerging evidence has indicated that some plant lncRNAs play multiple roles in the regulation of plant metabolism, development, stress responses, and other biological processes (Di et al. 2014; Shafiq et al. 2016; Wang et al. 2014). Numerous lncRNAs have been reported to participate in the responses to a wide variety of stresses in Arabidopsis,maize, medicago, and wheat (Liu et al. 2012; Wang et al. 2015b; Xin et al. 2011; Zhang et al. 2014). In addition, several stress-responsive lncRNAs such as COLDAIR, lncRNA16397, and Cdc28 have been functionally characterized in plants (Cui et al. 2016; Heo and Sung 2011; Nadal-Ribelles et al. 2014). However, we still have only a rudimentary understanding of the functions of most lncRNAs in response to stress.
Circular RNAs (circRNAs) are another categary of noncoding RNAs. They are circular in structure and are widespread in viroids or eukaryotes (Capel et al. 1993; Cocquerelle et al. 1993). CircRNAs can arise from exons or introns but are distinct structures with an independent manner of regulation (Memczak et al. 2013). Similar to lncRNAs, circRNAs show low expression levels and cell-type-specific patterns in eukaryotic cells (Salzman et al. 2013; Zuo et al. 2016). CircRNAs were suggested to be functionless when they have first identified in junk DNA some two decades ago (Nigro et al. 1991). However, recent studies have shown that circRNAs potentially play a role in enhancing the transcriptional activity of host genes (Hansen et al. 2013; Li et al. 2015; Memczak et al. 2013). Analyses of the expression patterns and regulatory functions of circRNAs have revealed that they are expressed in a range of eukaryotic organisms and have a regulatory pattern independent of, and different from, that of their cognate linear isoforms.
Stress-responsive lncRNAs and circRNAs have not been well studied in soybean. Little is known about how noncoding RNAs are expressed and responsible for gene expression regulation in soybean at the transcriptional or post-transcriptional levels during survival or acclimation responses. In this study, therefore, we conducted awhole-transcriptome analysis, identified comprehensive sets of soybeanlncRNAs and circRNAs, assessed the expression profiles of lncRNAs, and analyzed the regulatory mechanisms of lncRNAsunder drought and saline-alkaline stresses.
MATERIALS AND METHODS
Materials, stress treatments, and determination of physiological indexes: Seeds of the cultivated soybean variety “JY72” (a landrace cultivated in northeastern China) were placed in a sterile culture container in 1× Hoagland’s solution (containing 4 mL/L Fe sequestrene, 6 mM K+, and 4 mM Ca2+). At the stage of four leaves, the plants were subjected to abiotic stress for 24 hours under continuous light (Liu et al. 2012). To impose the drought and saline-alkaline stresses, the seedlings were incubated in liquid Hoagland’s medium containing 8% PEG and 110 mM NaCl + 50 mM NaHCO3, respectively. Seedlings in liquid Hoagland’s medium served as the control. The leaves were harvested from three groups per treatment, frozen with liquid nitrogen, and then stored at −80°C. Leaves were collected at 0 h, 6 h,12 h, and 24 h of the treatments to analyze superoxide dismutase (SOD) activity and malondialdehyde (MDA) content. A Tecan infinite M1000 Pro Microplate Reader(San Jose, CA, USA) was used to measure the absorbance of reaction solutions to determine SOD activity and calculate MDA content. The reaction solutions were prepared according to the instructions of a physiological assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). All the processes were biologically and temporally repeated in three independent and parallel experiments.
Library construction for RNA-seq and sequencing procedures: mirVana™ miRNA Isolation Kit (Ambion, Foster City, CA, USA) was employed to isolate total RNAs. The TruSeq® Stranded RNA Sample Preparation reagent (Illumina, San Diego, CA, USA) was used to prepare Strand-specific libraries by following the manufacturer’s instructions. We used Ribo-Zero rRNA removal beads to get rid of Ribosomal RNA from total RNA. After purification, total RNA was break up in to small pieces of fragments by using divalent cations at 95°C for 8 min. These RNA fragments were then synthesized into cDNA by using random primers. DNA Polymerase I was used to synthesis second-strand cDNA. Then, these cDNA fragments were precessed with end-repairment, including the addition of a single “A” base, and then ligated with adapters. Qubit® 2.0 Fluorometer (Life Technologies, Gaithersburg, MD,USA) was employed to quantify purified libraries and validated by Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) to make sure the insert size and measure concentration . Clusters were generated by cBot with the library diluted to 10 pM and sequencing by Illumina HiSeq 2500 sequencer (Illumina, USA).
Bioinformatics analysis: Atfer filtering rRNAs, adapters and low-quality reads, and short-fragments, FASTQC tools (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) were adopted to propress clean reads. We used TopHat v2.0.9 (Trapnell et al. 2009) to map the clean reads to the Glycine max v2.0 reference genome with two mismatches allowed. The transcriptomes of three libraries were assembled separately using Cufflinks and the whole genome sequence as a reference. by using Cuffcompare, when after comparing with the assembled transcript isoforms and the known protein coding transcripts, novel transcripts were gained. Putative lncRNAs were identified as novel transcripts, with the filters as followed: length more than 200 bp; exons more than 2; and ORF less than 300 bp (He et al. 2015; Wang et al. 2018; Wang et al. 2017); weak or non protein-coding ability (CPC score < 0 (Kong et al. 2007), CNCI score < 0 (Sun et al. 2013)), and no significant similarity with Pfam in a protein-coding database (Punta et al. 2012). We selected dynamicaly expressed lncRNAs used for predicting targets. The cis-acting target genes were considered as transcribed upstream or downstream of the lncRNAs within 10 kb distance. RNAplex software was emplpyed to predicted trans-acting targets.
We used CIRI version 1.2 (Gao et al. 2015) to characterize circRNAs. We used CIRI V1.2 to detect junction sites in sequence reads with PCC signals and PEM signals that reflected circRNA candidates by SAM alignment. Preliminary filtering was implemented using GT–AG splicing signals for the junction reads. After junction clustering, CIRI V1.2 was used to scan the SAM alignment again to detect junction-related reads and conduct further filtering to eliminate false-positive candidates.
qRT-PCR analyses: cDNA first strand was synthesized by using Reverse Transcript reagent (Takara, Dalian, China) with total RNA as template. All qRT-PCR reactions were operated on a Stratagene Mx3000P equipment (Agilent, USA) with a reaction volume of 25 µl consisting of 12.5 µl 2× SYBR premix Ex taq™ (Takara, Otsu, Japan), 2 µl cDNA and 10 µM specific primers. The soybean tubulin gene was employed as an internal control. 2−ΔΔCt formula was used to calculate the relative gene expression.
Data analysis: Data was subjected to a one-way ANOVA or Student’s t test to determine the statistical significance. SPSS was used for statistical analysis. Differences among groups were considered statistically significant at P < 0.05.
RESULTS
Identification of lncRNAs and circRNAs in soybean: The SOD activity and MDA content were significantlyincreased by 12 h of the drought and saline-alkaline treatments (Figure 1), confirming that the physiological status of these plants was seriously affected under these conditions. Therefore, plants at 12 hours of these stress treatments were used as the source of RNAs for library construction and sequencing (Table S1). A total of 119,631 transcripts were acquired from the three libraries (Control, drought stress (8% PEG-treated for 12 h), and saline-alkaline stress (110 mM NaCl + 50 mM NaHCO3-treatedfor 12 h). The assembled transcripts showing alignment (P value <1.0E−10, identity more than 90%, coverage more than 80%) to known soybean lncRNA transcripts significantly were identified as known lncRNAs (n=1,979, 47.81%) (NCBI Glycine_max_v2.0). The remaining transcripts were processed by size selection to identify novel lncRNAs. A total of 155 transcripts with length more than two hundreds nucleotides and with an ORF length of fewer than one hundred amino acids was extracted. After filtering based on exon number, 103 transcripts with two or more exons were selected. Finally, 41 novel lncRNAs were identified (Table S2). Of these, four were intronic lncRNAs, 28 were intergenic lncRNAs, and the remaining nine were antisense lncRNAs. We have supplied an annotation file of novel lncRNAs (Table S2) listing distinct lncRNAs that could be investigated by other researchers. A total of 2,020 lncRNAs were selected for further analysis. We compared the transcript length between lncRNAs and coding genes (Figure 2A). The transcript lengths of most lncRNAs were shorter than mRNAs, and fewer lncRNAs than mRNAs contained two or three exons (Figure 2B). Our analyses uncovered a set oflncRNAs expressed during the stress response in soybean. These results highlighted genes that may play functional roles in the stress response of soybean.

Figure 1.Superoxide dismutase activity and malondialdehyde content in soybean under drought and saline–alkaline stresses. * p < 0.05, **p <0.01
We also identified circRNAs from our dataset of mapping reads. We identified 830 non-redundant circRNAs, comprising 327, 452, and 353 candidate circRNAs that were recognized from junction reads in the datasets of Control, drought-, and saline–alkaline treated samples, respectively (Table S3). Most of the circRNAs were located in exonic and intergenic regions (Figure 2C). The mapping of junction reads showed that they were uniformly distributed among all the chromosomes (Figure 2D). . We detected circRNAs with different numbers of exons and from various intergenic regions. This may be attributed to their transcription sites in the genome or different regulatory functions.
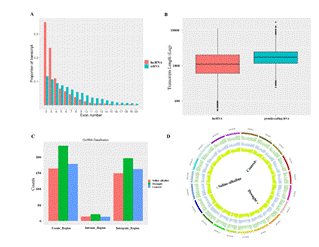
Figure 2. Identification of lncRNAs and circRNAs. a Number of exons in lncRNAs and coding RNAs. b Transcript levels of lncRNAs and coding RNAs. c Relative amount of each type of circRNAs. d Junction reads representing circRNAs (red) and mapping reads across treatment libraries mapped to genome.
Expression profiles of lncRNA: We determined the numbers of lncRNAs in the three libraries. In total, 1,765 (87.38%) of the lncRNAs were co-expressed among the control, drought, and saline-alkaline treatments.Interestingly, few lncRNAs were commonly expressed between control and the drought treatment, between control and the saline-alkaline treatment, or between the drought and saline-alkaline treatments. Only a few lncRNAs were explicitly expressed in the drought and saline-alkaline treatments (Figure 3A), implying that these lncRNAs were conserved regulators of plant development or the stress response.
To identify drought and saline-alkaline stress-responsive lncRNAs, the normalized expression FPKM(fragments per kilobase of transcript, per million fragments sequenced) number of lncRNAs were compared among the three libraries. Among the 2,020 lncRNAs, 143 were identified as differentially expressed. Of them, 118 exhibited changes in transcript levels under drought stress and 47 did so under saline-alkaline stress(Figure 3B). In the drought treatment, 109 (71.19%) lncRNAs were significantly highly expressed and 9 (28.81%) lncRNAs were down-regulated, as compared with control. In the saline-alkalinetreatment, 25 (53.19%) lncRNAs were up expressed and 22 (46.81%) lncRNAs were down-regulated, as compared with control. In addition, 10 co-expressedlncRNAs were up-regulated under both stresses, and 3 co-expressedlncRNAs were down-regulated under both stresses.
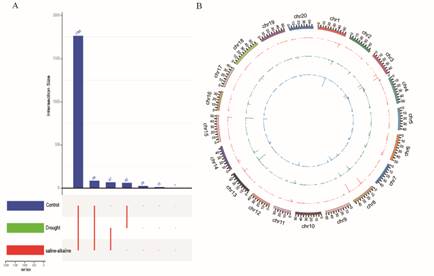
Figure 3. LncRNA expression profiles. a Categories of lncRNAs expressed under control, drought, and saline–alkaline conditions.b Relative abundance oflncRNA transcript levels under control (a, blue), drought (b, green), and saline-alkalineconditions (c, red).
Analyses of functional roles of potential targets oflncRNAs: The lncRNAs can take part in transcription process both in cis and in trans. Therefore, to predict the biological functions of genes affected by the differentially expressed lncRNAs underdrought and saline-alkalinestresses, we identified the mRNAs potentially regulated by the 143 differentially expressed lncRNAs incis or in trans.In this way, wepredicted 5,575 mRNA target genesof lncRNAs.
First, we analyzed the genes whose transcription could be regulated by lncRNAsin cis. We conducted Gene Ontology (GO) analyses of the target genes to assign functional categories. Comparing the drought treatment with control, the targets of the dynamically expressed lncRNAs were significantly enriched in the categories of nucleoplasm (p-value = 5.60e-04) and transcription from RNA polymerase II promoter (p-value = 1.31e-03).Comparing the saline-alkaline treatment with control, the targets of the 47 dynamically expressed lncRNAs were significantly enriched in the categories of transcription factor activity (p-value = 9.55e-03), and signal transduction (p-value = 1.36e-02)(Table S4).
We conducted the same analyses for the genes whose transcription could be regulated by lncRNAs in trans. The trans targets of the lncRNAs differentially expressed under drought were significantly enriched in biological processes, including ATPase activity (p-value =1.47e-03), r pyrophosphatase activity (p-value = 1.29e-03), and acting on acid anhydrides (p-value = 1.35e-03).In th saline-alkaline treatment, the trans targets of the differentiallyexpressedlncRNAs were significantly enriched in molecular functions, including hydrolyzing O-glycosyl compounds (p-value = 1.85e-04), acting on glycosyl bonds (p-value = 3.00e-04), and carbohydrate metabolic process (p-value = 2.47e-03). These results indicated that specific genes and pathways were regulated by differentially expressed lncRNAs during the plant stress response.
Together, these results showed that the lncRNAs whose expression levels changed upon exposure to drought and saline-alkaline stresses were functionally significant in pathways via their regulation of RNA processing ina cis- or trans-acting manner.
Expression validation of lncRNAs and targets: We selected 8 pairs of lncRNAs and the potential target mRNAsto validate their expression pattern under stresses by qRT-PCR. Excepted two pairs of lncRNA and potential targets (XR_001386720.1/LOC100792149 and XR_419416.2/LOC100815558), the others showed consistency with sequencing results (Figure 4).

Figure 4.Expression validation of candidatelnRNAs and potential targets.
Characterization and validation of circRNAs: Soybean circRNAs generated from exonic, intergenic, and intronic regions were identified according to the transcriptomic data.Among the identified circRNAs, 123 were generated from exons of a single protein-coding gene and were classified as exoniccircRNAs, and 223 were generated from intergenic regions. There was a relatively low rate of circRNAs from the intron-flanking regions. Splicing junction sites are one of the most important factors delineating the origin of the circRNAs and in differentiating linear splicing from back-splicing. We found that seven of 12 circRNAs (four exoniccircRNAs, two intronic circRNA, and four intergenic circRNAs) had junction sites (Figure 6a and Figure S1). Primers were specifically designed for circRNA segment amplification (Figure 6b, c, d). The expression patterns of some circRNAs were verified by qRT-PCR (Figure 6e). Only circ_LOC100818869 was expressed at low levels under stress conditions. All the other circRNAs were expressed at high levels under stress conditions.
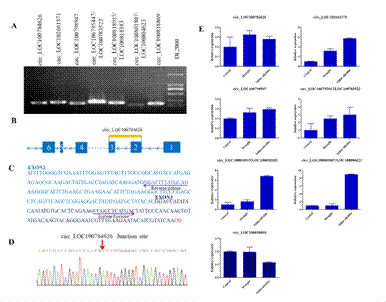
Figure 5. Characterization and validation of circRNAs. a PCR product amplified with lncRNA-specific primers from soybean cDNA. b Distribution of circ_LOC100784626 in cullin-1 exon. c Primers designed to amplify circ_LOC100784626. d Sequencing results showing junction sites in circ_LOC100784626. e Expression of circRNA validated by qRT-PCR. * p < 0.05, ** p < 0.01
DISCUSSION
Noncoding RNAs such as lncRNAs have been detected out or identified by many different techniques, such as strand-specific microarrays, conventional expression arrays, next generation RNA sequencing, or real-time experiments (Herzog et al. 2014; Lu et al. 2012). Strand-specific next generation RNA sequencing permitred the recovery of antisense transcription activities and led to novel lncRNAs recognition. At present, plant biologists utilized this method to analyze transcriptomes and characterize a wide range of noncoding RNAs in many plant varieties (Cruz de Carvalho et al. 2016; Joshi et al. 2016; Wang et al. 2015a). Building on this work, we performed genome-wide identification of noncoding RNAs by analyzing strand-specific paired-end RNA sequencing data. A total of 2,020 lncRNAs and 830 circRNAs were identified from the strand-specific RNA sequencing data. We found far fewer lncRNAs than were found in another recent study (Li et al. 2014), possibly because we used more rigorous filtering criteria to identify lncRNAs.
The expression profiles of circRNAs were not considered in this study. After discarding low-mapping-quality reads and back-spliced junction sequences with homology to linear exon junctions, most of the identified circRNAs showed very low expression levels and high false-positive rates. Althoughnew methods such as CIRI-full have been proposed to evaluate transcript levels of circRNAs(Zheng et al. 2019), a comprehensive analysis of circRNA expression is still lacking.
We focused on the expression patterns and features of lncRNAs in this study. To understand the tolerance mechanisms of soybean, we first profiled the drought and saline-alkaline induced changes in the expression profiles of lncRNAs. We found 118 and 47 differentially expressed lncRNAs in response to dehydration or saline-alkaline conditions at the transcriptional level. According to recent studies, lncRNAs primarily regulate transcription in a cis- or trans- manner to positively or negatively control the expression of coding genes (Bumgarner et al. 2009; Guil and Esteller 2012). In our study, we found that lncRNAs targeted 5,575 mRNAs in acis- or trans- manner, indicating that there arewidespread regulatory interactions between lncRNAs and coding RNAsin soybean. In the drought and saline-alkaline treatments, the regulation of cell signaling was affected by 6 and 8 differentially expressed lncRNAs, respectively. The putative targets of the dynamically expressed lncRNAs with cis-acting regulation included genes encoding auxin response factor 3 (ARF3), IAA, and ras-related protein (RABA1). These findings suggested that lncRNAs are pariticipated in regulating the expression of RNAs involved in signal transduction at the transcriptional level. This conclusion is consistent with the recent finding that lncRNAs function as regulators of cell signaling under stress (Saeedi Borujeni et al. 2018). Previous analyses have revealed that certain lncRNAs may improve plants’ resistance to stress (Liu and Zhu 2014).Here, we identified differentially expressedlncRNAs under drought and saline–alkaline stresses.As an intergenic lncRNA, XR_001389476.1 regulates the expression of a gene encoding an F-box protein in acis-acting manner in soybean. Drought caused a rapid change in the XR_001389476.1 expression level. Previous studies have shown that the F-box gene expression level increases underoxidative stress and affects the levels of signaling molecules such as reactive oxygen species (Kong et al. 2016). Further research is needed to elucidate the molecular mechanism of lncRNA in soybean.
CircRNAs are produced by head-to-tail back-splicing events and are participated in the RNA expression regulation at the translational level or the post-translational level (Darbani et al. 2016). CircRNAs was recently reported to be novel regulators of gene expression in plants (Zuo et al. 2016). Using strand-specific RNA-seq methods, we identified 830 circRNAs originating from diverse genomic regions under our stress conditions. Despite the low abundance of circRNAs, we validated the back-splicing sites to confirm the existence of circRNAs generated from exonic, intronic, and intergenic regions (Siegel et al. 2014). Taken together, our research indicated that circRNAs may have additional functions in plants and/or undiscovered functions in stress responses.
Conclusion: In summary, with the aid of strand-specific transcriptomic analysis, we identified 2,020 lncRNAs, including 1,979 known and 41 novel ones, in soybean. In addition, 830 circRNAs were identified. The results of GO functional enrichment analyses indicated that the dynamically expressed lncRNAs were paricipitated in the regulation of certain biological processes. Putative trans/cis targets of the dynamically expressed lncRNAs were predicted. Most circRNAs originated from exonic, intronic, and intergenic sequences of G. max and back-spliced junction sites of predicted circRNAs were validated through Sanger sequencing. These results increase our knowledge of the lncRNAs and circRNAs that are involved in soybean stress responses.
Acknowledgements: This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFD0101005), the Special Program for Research of Transgenic Plants(Grant No. 2016ZX08010002-004), the National Natural Science Foundation of China (Grant No. 31601323, 31771868), and the 13th Five-year Plan of Science and Technology Project of Jilin(Grant No.JJKH20190918KJ).
REFERENCES
- Bouain, N., G. Krouk, B. Lacombe and H. Rouached. (2019). Getting to the Root of Plant Mineral Nutrition: Combinatorial Nutrient Stresses Reveal Emergent Properties. Trends in plant science 24(6). 542-552.
- Bumgarner, S.L., R.D. Dowell, P. Grisafi, D.K. Gifford and G.R. Fink. (2009). Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proceedings of the National Academy of Sciences 106(43). 18321-18326.
- Capel,, A. Swain, S. Nicolis, A. Hacker, M. Walter, P. Koopman, P. Goodfellow and R. Lovell-Badge. (1993). Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 73(5). 1019-1030.
- Cocquerelle C., B. Mascrez, D. Hetuin and B. Bailleul. (1993). Mis-splicing yields circular RNA molecules. FASEB J 7(1).155-160.
- Cruz de Carvalho, M.H., H.X. Sun, C. Bowler and N.H. Chua. (2016). Noncoding and coding transcriptome responses of a marine diatom to phosphate fluctuations. New Phytol 210(2). 497-510.
- Cui, J., Y. Luan, N. Jiang, H. Bao and J. Meng . (2016). Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J 89(3). 577-589.
- Darbani,, S. Noeparvar and S. Borg. (2016). Identification of Circular RNAs from the Parental Genes Involved in Multiple Aspects of Cellular Metabolism in Barley. Frontiers in plant science 7. 776.
- Deinlein,, A.B. Stephan, T. Horie, W. Luo, G. Xu and J.I. Schroeder. (2014). Plant salt-tolerance mechanisms. Trends in plant science19(6).371-9.
- Di,, J. Yuan, Y. Wu, J. Li, H. Lin, L. Hu, T. Zhang, Y. Qi, M.B. Gerstein, Y. Guo and Z.J. Lu. (2014). Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J80(5).848-61.
- Gao,, J. Wang and F. Zhao. (2015). CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16(1).4.
- Guil, and M. Esteller. (2012). Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol 19(11). 1068-1075.
- Hansen,B., T.I. Jensen, B.H. Clausen, J.B. Bramsen, B. Finsen, C.K. Damgaard and J. Kjems. (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495(7441).384-388.
- He,, Q. Liu, L. Zheng, Y. Cui, Z. Shen and L. Zheng. (2015). RNA-Seq Analysis of Rice Roots Reveals the Involvement of Post-Transcriptional Regulation in Response to Cadmium Stress. Frontiers in plant science 6. 1136.
- Heo,B. and S. Sung. (2011). Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 331(6013).76-9.
- Herzog,A., A. Lempradl, J. Trupke, H. Okulski, C. Altmutter, F. Ruge, B. Boidol, S. Kubicek, G. Schmauss, K. Aumayr, M. Ruf, A. Pospisilik, A. Dimond, H.B. Senergin, M.L. Vargas, J.A. Simon and L. Ringrose. (2014). A strand-specific switch in noncoding transcription switches the function of a Polycomb/Trithorax response element. Nat Genet 46(9).973-981.
- Joshi,K., S. Megha, U. Basu, M.H. Rahman and N.N. Kav. (2016). Genome Wide Identification and Functional Prediction of Long Non-Coding RNAs Responsive to Sclerotinia sclerotiorum Infection in Brassica napus. PLoS One 11(7). e0158784.
- Kapranov,, J. Cheng, S. Dike, D.A. Nix, R. Duttagupta, A.T. Willingham, P.F. Stadler, J. Hertel, J. Hackermuller, I.L. Hofacker, I. Bell, E. Cheung, J. Drenkow, E. Dumais, S. Patel, G. Helt, M. Ganesh, S. Ghosh, A. Piccolboni, V. Sementchenko, H. Tammana and T.R. Gingeras. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316(5830).1484-1488.
- Kong,, Y. Zhang, Z.Q. Ye, X.Q. Liu, S.Q. Zhao, L. Wei and G. Gao. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35. W345-349.
- Kong,, S. Zhou, S. Yin, Z. Zhao, Y. Han and W. Wang. (2016). Stress-Inducible Expression of an F-box Gene TaFBA1 from Wheat Enhanced the Drought Tolerance in Transgenic Tobacco Plants without Impacting Growth and Development. Frontiers in plant science 7. 1295.
- Kunert,J., B.J. Vorster, B.A. Fenta, T. Kibido, G. Dionisio and C.H. Foyer. (2016). Drought Stress Responses in Soybean Roots and Nodules. Frontiers in plant science 7. 1015.
- Kung,T., Colognori D and Lee JT. (2013). Long noncoding RNAs: past, present, and future. Genetics 193. 651-669.
- Li,, B. Wu, J. Xu and C. Liu. (2014). Genome-wide identification and characterization of long intergenic non-coding RNAs in Ganoderma lucidum. PLoS One 9. e99442.
- Li,, C. Huang, C. Bao, L. Chen, M. Lin, M. Wang M, G. Zhong, B. Yu, W. Hu, L. Dai, P. Zhu, Z. Chang, Q. Wu, Y. Zhao, Y. Jia, P. Xu, H. Liu and G. Shan. (2015). Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22(3). 256-264.
- Liu,, C. Jung, Xu J, H. Wang, S. Deng, L. Bernad, C. Arenas-Huertero and N.H. Chua. (2012). Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 24(11). 4333-4345.
- Liu, and J-K. Zhu. (2014). Non-coding RNAs as potent tools for crop improvement. National Science Review 1. 186-189.
- Liu,, Hao L, Li D, Zhu L and Hu S. (2015). Long non-coding RNAs and their biological roles in plants. Genomics Proteomics Bioinformatics 13(3). 137-147.
- Lu,, C. Zhu, G. Lu, Y. Guo, Y. Zhou, Z. Zhang, Y. Zhao, W. Li, Y. Lu, W. Tang, Q. Feng and B. Han. (2012). Strand-specific RNA-seq reveals widespread occurrence of novel cis-natural antisense transcripts in rice. BMC genomics 13. 721.
- Memczak,, M. Jens, A. Elefsinioti, F. Torti, J. Krueger, A. Rybak, L. Maier, S.D. Mackowiak, L.H. Gregersen, M. Munschauer, A. Loewer, U. Ziebold, M. Landthaler, C. Kocks, F. le Noble and N. Rajewsky. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441). 333-338.
- Nadal-Ribelles,, C. Sole, Z. Xu, L.M. Steinmetz, E. de Nadal and F. Posas. (2014). Control of Cdc28 CDK1 by a stress-induced lncRNA. Mol Cell 53(4). 549-561.
- Nigro J.M., K.R. Cho, E.R. Fearon, S.E. Kern, J.M. Ruppert, J.D. Oliner, K.W. Kinzler and B. Vogelstein. (1991). Scrambled exons. Cell 64(3). 607-613.
- Punta,, P.C. Coggill, R.Y. Eberhardt, J. Mistry, J. Tate, C. Boursnell, N. Pang, K. Forslund, G. Ceric, J. Clements, A. Heger, L. Holm, E.L. Sonnhammer, S.R. Eddy, A. Bateman and R.D. Finn. (2012). The Pfam protein families database. Nucleic Acids Res 40. D290-301.
- Qi,, S. Xie, Y. Liu, F. Yi and J. Yu. (2013). Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol Biol 83(4-5). 459-473.
- Rivas E., J. Clements and S.R. Eddy. (2017). A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods 14(1). 45-48.
- Rymarquis,A., J.P. Kastenmayer, A.G. Huttenhofer and P.J. Green. (2008). Diamonds in the rough: mRNA-like non-coding RNAs. Trends in plant science 13(7). 329-334.
- Saeedi Borujeni,J., E. Esfandiary, A. Baradaran, A. Valiani, M. Ghanadian, P. Codoner-Franch, R. Basirat, E. Alonso-Iglesias, H. Mirzaei and A. Yazdani. (2018). Molecular aspects of pancreatic beta-cell dysfunction: Oxidative stress, microRNA, and long noncoding RNA. Journal of cellular physiology.234(6).8411-8425.
- Salzman,, R.E. Chen, M.N. Olsen, P.L. Wang and P.O. Brown. (2013). Cell-type specific features of circular RNA expression. PLoS Genet 9(9). e1003777.
- Shafiq,, J.Li and Q. Sun. (2016). Functions of plants long non-coding RNAs. Biochim Biophys Acta 1859(1). 155-162.
- Siegel,N., C.C. Hon, Q. Zhang, J.J. Lopez-Rubio, C. Scheidig-Benatar, R.M. Martins, O. Sismeiro, J.Y. Coppee and A. Scherf. (2014). Strand-specific RNA-Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum. BMC genomics 15(1). 150.
- Sun,, H. Luo, D. Bu, G. Zhao, K. Yu, C. Zhang, Y. Liu, R. Chen and Y. Zhao. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41(17). e166.
- Trapnell,, L. Pachter and S.L. Salzberg. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9). 1105-1111.
- Wang,, J. Lin, J. Kan, H. Wang, X. Li, Q. Yang, H. Li and Y. Chang. (2018). Genome-Wide Identification and Functional Prediction of Novel Drought-Responsive lncRNAs in Pyrus betulifolia. Genes 9(6).311
- Wang,, W. Yu, Y. Yang, X. Li, T. Chen, T. Liu, N. Ma, X. Yang, R. Liu and B. Zhang. (2015a). Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci Rep 5. 16946.
- Wang,, W. Ge, Z. Luo, Y. Guo, B. Jiao, L. Qu, Z. Zhang and X. Wang. (2017). Integrated analysis of coding genes and non-coding RNAs during hair follicle cycle of cashmere goat (Capra hircus). BMC genomics 18(1). 767.
- Wang,Z., M. Liu, M.G. Zhao, R. Chen and W.H. Zhang. (2015b). Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol 15. 131.
- Wang,, X. Wang, W. Deng, X. Fan, T.T. Liu, G. He, R. Chen, W. Terzaghi, D. Zhu and X.W. Deng. (2014). Genomic features and regulatory roles of intermediate-sized non-coding RNAs in Arabidopsis. Mol Plant 7. 514-527.
- Xin,, Y. Wang, Y. Yao, N. Song, Z. Hu, D. Qin, C. Xie, H. Peng, Z. Ni, and Sun Q. (2011). Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol 11. 61.
- Zhang,, Z. Han, Q. Guo, Y. Liu, Y. Zheng, F. Wu and Jin W. (2014). Identification of maize long non-coding RNAs responsive to drought stress. PLoS One 9(6). e98958.
- Zheng,, Ji P, S. Chen, L. Hou and Zhao F. (2019). Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome medicine 11(1). 2.
- Zuo,, Q. Wang, B. Zhu, Y. Luo and Gao L. (2016). Deciphering the roles of circRNAs on chilling injury in tomato. Biochem Biophys Res Commun 479(2). 132-138.
|





